VASP计算钻石带隙温度依赖性的完整教程

1. 带隙温度依赖性计算概述
在材料计算领域,研究带隙随温度变化的特性对于理解半导体材料的光电性能至关重要。钻石作为一种典型的宽禁带半导体,其带隙的温度依赖性一直是研究热点。VASP(Vienna Ab initio Simulation Package)作为第一性原理计算的利器,提供了计算带隙温度依赖性的完整解决方案。
这个教程的核心在于利用单次方法(one-shot method)计算钻石的零点重整化(Zero-Point Renormalization, ZPR)和间接带隙的温度依赖性。这种方法通过考虑晶格振动对电子结构的影响,能够更准确地预测材料在不同温度下的带隙变化。
提示:在实际研究中,带隙的温度依赖性主要来自两个方面的贡献:电子-声子耦合效应和热膨胀效应。本教程首先关注电子-声子耦合,后续会引入体积效应进行更精确的计算。
2. 计算准备与超胞构建
2.1 初始结构准备
计算从钻石的原始晶胞开始,其POSCAR文件如下:
bash复制C_2_fcc
1.00000000000000
0.0000000000000000 1.783493 1.783493
1.783493 0.0000000000000000 1.783493
1.783493 1.783493 0.0000000000000000
C
2
Direct
0.0000000000000000 0.0000000000000000 0.0000000000000000
0.7500000000000000 0.7500000000000000 0.7500000000000000
这个结构描述了一个包含2个碳原子的面心立方晶胞,晶格常数为3.567Å(1.783493×2)。
2.2 超胞构建与收敛性测试
为了准确描述声子效应,需要构建足够大的超胞。官网教程使用4×4×4超胞(128个原子)进行演示,但实际研究中必须进行收敛性测试:
bash复制# 4×4×4超胞示例
C_128_fcc
1.00000000000000
0.00000000 7.13397200 7.13397200
7.13397200 0.00000000 7.13397200
7.13397200 7.13397200 0.00000000
C
128
Direct
[...128个原子坐标...]
实际操作中,建议依次计算3×3×3(54原子)、4×4×4(128原子)和5×5×5(250原子)超胞,观察带隙变化是否收敛。从示例结果看:
- 3×3×3超胞:ZPR = -0.258533 eV
- 4×4×4超胞:ZPR = -0.353883 eV
- 5×5×5超胞:ZPR = -0.304933 eV
当相邻尺寸超胞的结果差异小于0.05eV时,可认为已经收敛。
注意:超胞越大,计算量呈立方增长。需要在精度和计算成本间权衡,通常4×4×4超胞是较好的折中选择。
3. 零点重整化(ZPR)计算
3.1 计算流程详解
ZPR计算分为两个主要步骤:
- 初始计算:使用INCAR.init计算未考虑原子位移的电子结构
bash复制System = cd-C
PREC = Accurate
ALGO = FAST
ISMEAR = 0; SIGMA = 0.1;
IBRION = 6
PHON_LMC = .TRUE.
PHON_NSTRUCT = 0
PHON_TLIST = 0.0
- 位移结构计算:生成考虑量子涨落的位移结构并计算
bash复制# 生成位移结构
cp POSCAR.4x4x4 POSCAR
cp INCAR.init INCAR
# 运行VASP得到位移结构POSCAR.T=0.
# 自洽计算
cp POSCAR.T=0. POSCAR
cp INCAR.scf INCAR
# 运行VASP得到OUTCAR.T=0.
3.2 结果提取脚本解析
使用extract_zpr.sh脚本提取ZPR结果:
bash复制#!/bin/bash
i="OUTCAR.T=0."
j="OUTCAR.init"
# 提取HOMO和LUMO能级
[...脚本内容...]
echo "The band gap (in eV) without zero-point vibrations is:"
echo "$h_ref $l_ref" |awk '{print ($2-$1)}'
echo "The band gap (in eV) including zero-point vibrations is:"
echo "$e1a $e1b $e1c $e2a $e2b $e2c $e2d $e2e $e2f" |awk '{print (($4+$5+$6+$7+$8+$9)/6.0-($1+$2+$3)/3.0)}'
echo "The zero-point renormalization of the band gap (in eV) is:"
echo "$e1a $e1b $e1c $e2a $e2b $e2c $e2d $e2e $e2f $h_ref $l_ref" |awk '{print (($4+$5+$6+$7+$8+$9)/6.0-($1+$2+$3)/3.0)-($11-$10)}'
脚本通过比较考虑和不考虑零点振动时的带隙差异,得到ZPR值。对于4×4×4超胞,典型结果为:
- 未考虑ZPR的带隙:4.4049 eV
- 考虑ZPR的带隙:4.05102 eV
- ZPR值:-0.353883 eV
4. 温度依赖性计算
4.1 多温度点计算设置
在INCAR.init中设置温度序列:
bash复制PHON_TLIST = 0.0 100.0 200.0 300.0 400.0 500.0 600.0 700.0
这将生成不同温度下的位移结构:
- POSCAR.T=0.
- POSCAR.T=100.
- ...
- POSCAR.T=700.
对每个温度结构进行自洽计算,得到对应的OUTCAR文件。
4.2 温度依赖数据提取
使用extract_temp.sh脚本处理多温度数据:
bash复制#!/bin/bash
[...脚本内容...]
for temp in 0 100 200 300 400 500 600 700
do
i="OUTCAR.T=$temp"
[...提取各温度带隙...]
echo "$temp $bandgap" >> gap_vs_temp.dat
done
4.3 结果可视化
使用gnuplot绘制带隙随温度变化曲线:
bash复制gnuplot -e "set terminal pngcairo size 800,600; set xlabel 'T (K)'; set ylabel 'Band gap (eV)'; set style data lines; plot 'gap_vs_temp.dat' title 'Calculated', 'C_exp_points_offset0.dat' w circles title 'Exp. data', 'C_exp_fit_offset0.dat' title 'Exp. fit'" > gap_vs_temp.png
典型结果如下图所示,计算值与实验数据吻合良好:
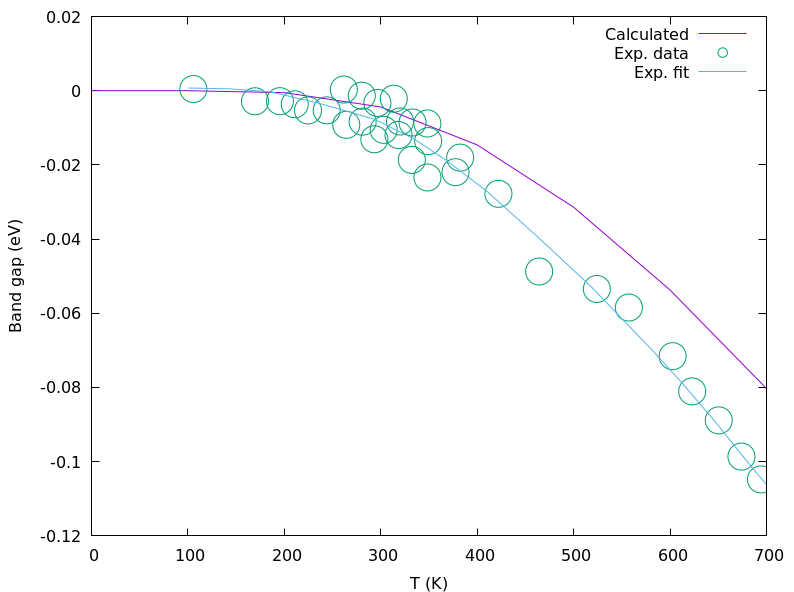
5. 考虑体积效应的精确计算
5.1 准谐近似计算
为获得更精确的结果,需要考虑热膨胀带来的体积效应。通过以下步骤实现:
- 在平衡体积±2%范围内生成15个不同体积的POSCAR
- 计算每个体积的动力学矩阵
- 拟合自由能-体积曲线
使用quasi_harm_4x4x4_diamond_create_pos_and_run_vasp.sh脚本:
bash复制#!/bin/bash
vasp_exec=./vasp_gam
np=8
cp INCAR.qh INCAR
for i in 6.13521592 6.27789536 6.4205748 6.56325424 6.70593368 6.84861312 6.99129256 7.133972 7.27665144 7.41933088 7.56201032 7.70468976 7.8473692 7.99004864 8.13272808
do
sed "s/7.13397200/${i}/g" POSCAR.4x4x4 > POSCAR_$i
cp POSCAR_$i POSCAR
mpirun -np $np $vasp_exec
mv OUTCAR OUTCAR_$i
done
5.2 自由能-体积曲线拟合
使用Birch-Murnaghan状态方程拟合每个温度下的平衡体积:
bash复制#!/bin/bash
for i in OUTTEMP_*
do
[...拟合过程...]
echo "temperature: $temp, a_latt: $a"
done
拟合结果示例:
code复制temperature: 0.00000000e+00, a_latt: 7.18018
temperature: 1.00000000e+02, a_latt: 7.18021
[...]
temperature: 7.00000000e+02, a_latt: 7.19976
5.3 最终温度依赖结果
更新晶格常数后重新计算温度依赖性,考虑体积效应后的结果与实验值符合得更好:
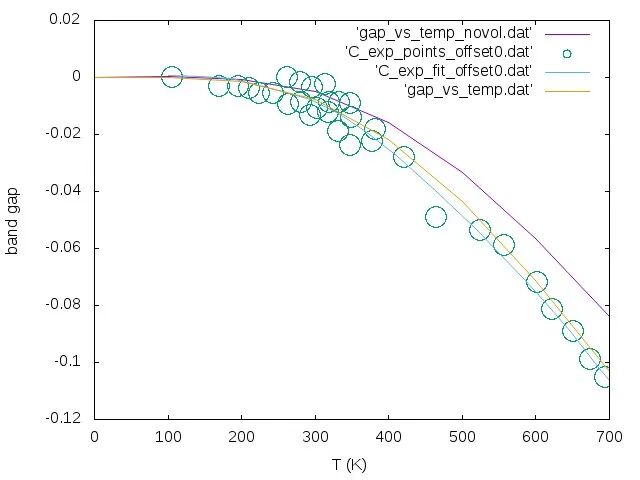
6. 常见问题与解决方案
6.1 计算不收敛问题
问题现象:电子自洽计算不收敛或声子计算报错。
解决方案:
- 检查POSCAR文件格式是否正确,特别是超胞构建后的原子坐标
- 调整INCAR中的参数:
- 增加ENCUT(通常设为1.3×POTCAR中的ENMAX)
- 调整SIGMA(金属用0.2,半导体/绝缘体用0.05-0.1)
- 使用ALGO = Normal替代FAST提高稳定性
6.2 内存不足问题
问题现象:计算大超胞时出现内存不足错误。
解决方案:
- 减少KPOINTS数量(对于超胞计算,Γ点通常足够)
- 在INCAR中设置KPAR = 2或4进行k点并行
- 使用NCORE参数优化核间通信(通常设为每节点物理核心数)
6.3 结果异常检查
问题现象:ZPR值异常大或温度趋势与预期不符。
排查步骤:
- 确认超胞尺寸是否足够(检查3×3×3、4×4×4、5×5×5结果是否收敛)
- 检查位移结构的幅度是否合理(查看POSCAR.T=*中的原子位移)
- 验证脚本提取的能级是否正确(手动检查OUTCAR中的能级分布)
7. 计算效率优化技巧
7.1 并行计算配置
对于大超胞计算,合理的并行设置可显著提高效率:
bash复制# 在提交脚本中设置
export OMP_NUM_THREADS=1
mpirun -np 32 vasp_std
对应INCAR设置:
bash复制NCORE = 4 # 每个组态计算使用的核心数
KPAR = 8 # 将k点分成8组并行
7.2 分步计算策略
为节省计算资源,可以采用分步策略:
- 先用小超胞(3×3×3)测试参数和脚本
- 确定参数后,再计算大超胞(4×4×4或5×5×5)
- 温度依赖性计算可先稀疏采样(如每200K一个点),再在关键区域加密
7.3 结果复用技巧
相同超胞尺寸的不同计算可以复用部分结果:
- 电子自洽计算的CHGCAR可用于初始化后续计算
- 声子计算的力常数矩阵可以用于多个温度点的位移生成
- 体积效应计算中,不同体积的结构可以并行提交
在实际操作中发现,使用4×4×4超胞在32核服务器上完成全套计算约需24-48小时,具体取决于系统规模和精度设置。建议先在小体系上验证整个流程,再开展大规模计算。